Abstract
GECCO is a precise and scalable method for the de novo identification of biosynthetic gene clusters (BGCs) in genomic and metagenomic data using Conditional Random Fields (CRFs).
Biosynthetic gene clusters (BGCs) are enticing targets for (meta)genomic mining efforts, as they may encode novel, specialised metabolites with potential uses in medicine and biotechnology. GECCO’s precision in identifying BGCs, including ones with novel architectures, has been extensively evaluated against large manually curated sets of BGCs. When applied to large genome collections, GECCO identified nearly twice as many BGCs compared to a rule-based approach, while achieving higher accuracy, in particular for the detection of BGC boundaries, than other machine learning and AI-based approaches.
The GECCO software is implemented in Python, supports all versions from Python 3.7 and is provided under the GNU General Public License v3.0 or later.
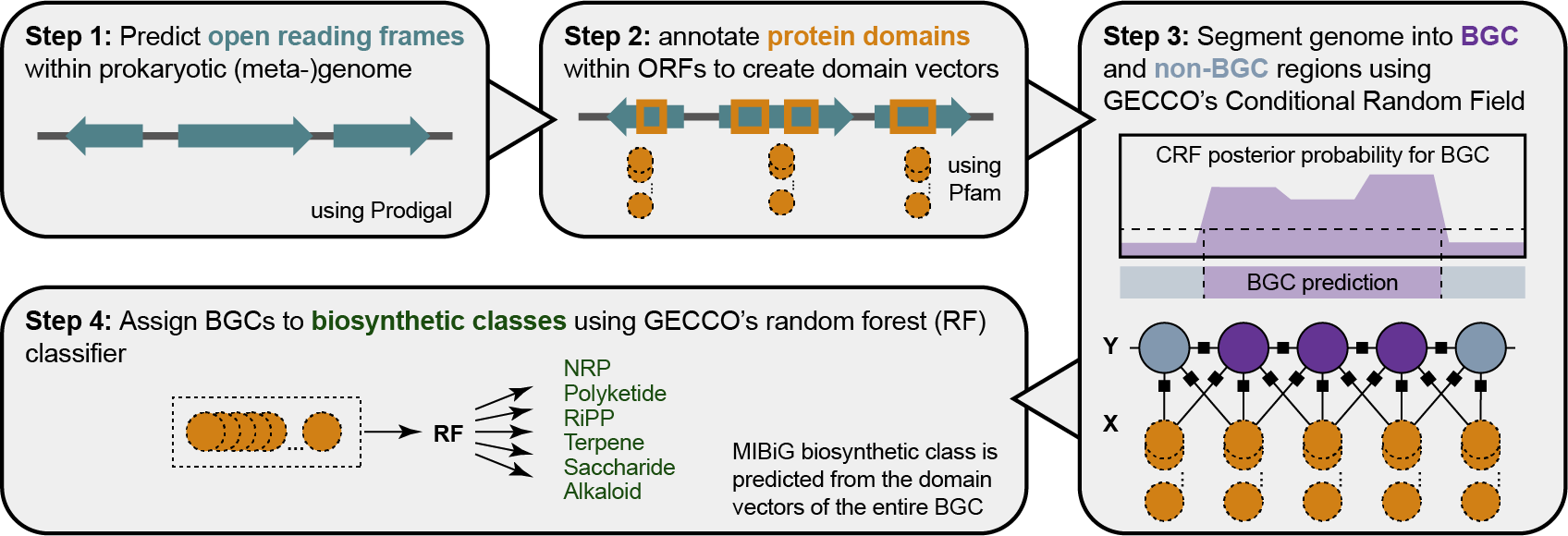
Graphical depiction of the biosynthetic gene cluster (BGC) identification and classification approach implemented in GECCO. Briefly, GECCO identifies open reading frames (ORFs) in an assembled prokaryotic (meta)genome (Step 1). Protein domains are annotated in the resulting ORFs using profile hidden Markov models (pHMMs; Step 2). The resulting ordered domain vectors serve as features for a conditional random field (CRF) that is trained to predict whether a stretch of contiguous genes belong to a BGC or not (Step 3). Predicted BGCs are classified into one of six major biosynthetic classes as defined in the Minimum Information about a Biosynthetic Gene cluster (MIBiG) database using a Random Forest classifier (Step 4).
GECCO has been cited in the following publications:
- MIBiG 3.0: a community-driven effort to annotate experimentally validated biosynthetic gene clusters
- Artificial intelligence for natural product drug discovery
- proGenomes3: approaching one million accurately and consistently annotated high-quality prokaryotic genomes
- Genome mining strategies for metallophore discovery